 New Products
New Products Earth-Friendly Products
Earth-Friendly Products Biotium Choice Antibodies
Biotium Choice Antibodies Special Offers
Special Offers







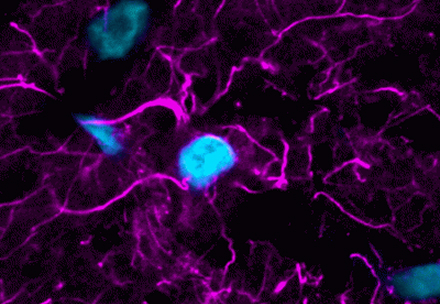
Content #1
Content #1
Content #1
In the 1670s, Dutch scientist Antonie Von Leeuwenhoek first discovered microbial life forms through his simple light microscope, a finding that would initiate a revolution in the field of microscopy.1 Since his early observations of what he termed ‘animacules’, the field has seen a quantum leap from his self-designed single lens microscopes to the current high resolution cryo-electron microscopes.2 Among these technological advances was the advent of fluorescence microscopy, a field that remains pivotal to modern biological research.3 In this article we will explore the basic principles governing fluorescence microscopes, the components involved, and its different variants.
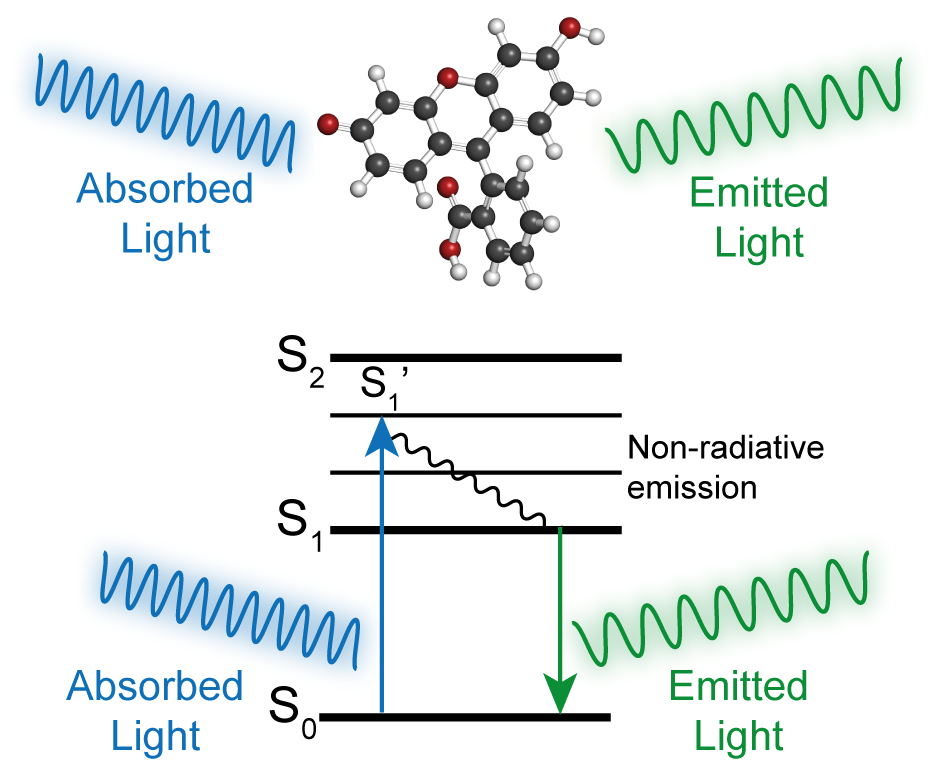
Figure 1. Jablonski Diagram of excitation and emission of a fluorescent dye.
It was not until the early 1900s that the principles of fluorescence was extrapolated to microscopy. The first working fluorescent microscope was developed by Oskar Heimstaedt in 1911. Initially, fluorescence microscopy was performed with transmitted light, with the path of the light beam following the design of a light microscope. The problem with this layout was that the emitted fluorescence is weak in comparison to the excitation light, resulting in masking the signals from the specimen. This issue was rectified by Philip Ellinger and August Hert in 1929 by placing the excitation and emission optics on the same side of the specimen, a method known as epi-illumination.1,4,6
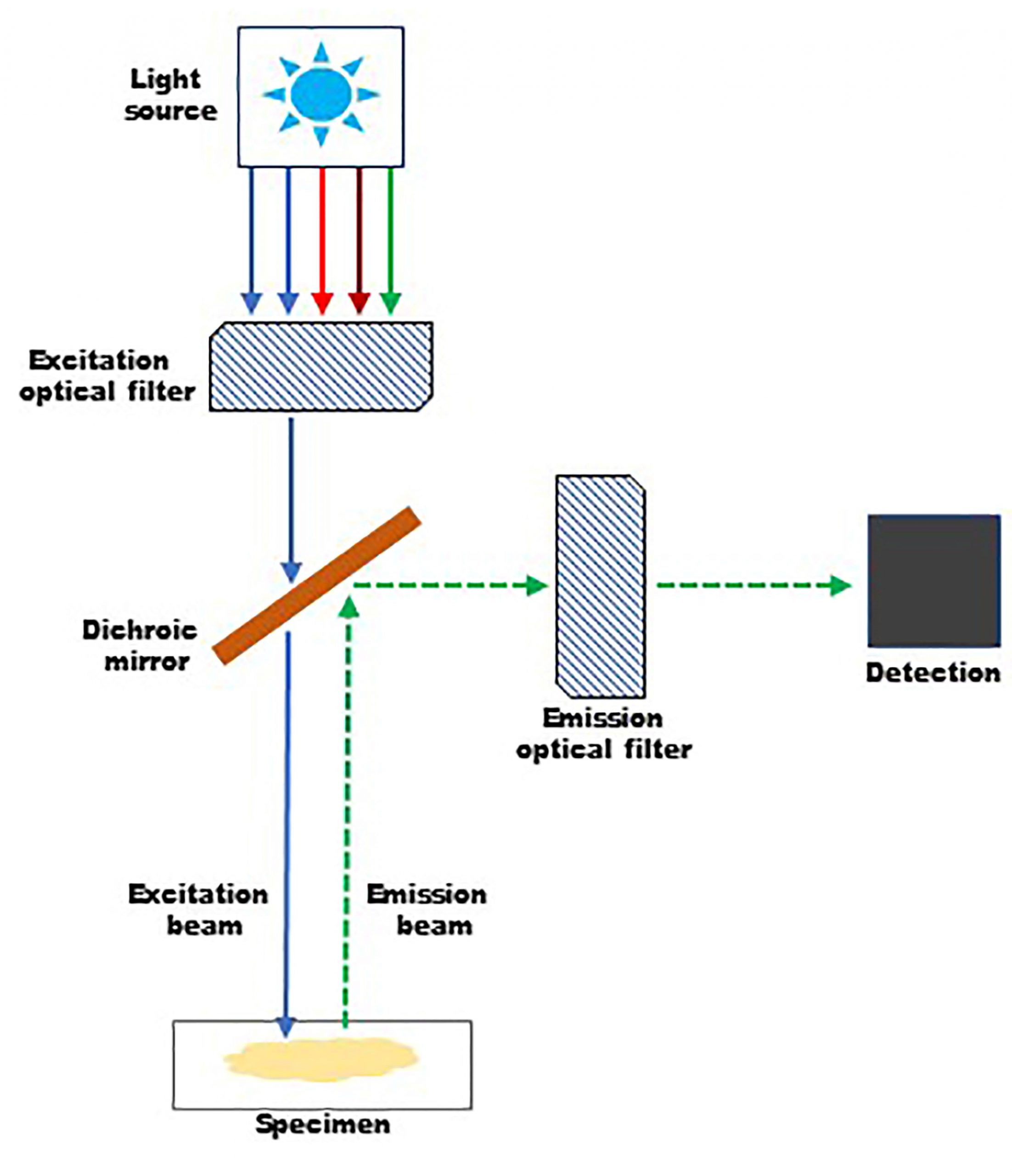
Figure 2. Basic design of a fluorescent microscope and epi-illumination.
Dyes, stains, or fluorescent proteins that label components of a specimen are known as fluorescence indicators or fluorophores. These fluorophores are the molecules that absorb and emit light at specific wavelengths to produce a fluorescent image. When multiple probes are required, careful selection of fluorophores with non-overlapping Stokes shift wavelengths are desired to minimize ‘bleed-through’ between signals.9
The basic fluorescence microscope offers a wide-field view with respect to specimen imaging. This broad illumination field lead to noise from regions outside the plane of focus (bidirectionally as in horizontal and vertical planes), leading to lower resolution. A simple solution for this was placing a pinhole between the path of both the excitation and emission light beams, a technology now known as confocal microscopy. Apart from superior resolution, confocal technology also facilitated the scanning of a specimen to reconstruct complex high-resolution three-dimensional images.10
Photobleaching occurs when fluorophores are photochemically altered by the excitation source such that the molecule is unable to fluoresce. This is particularly pertinent to laser scanning confocal microscopes due to the high energy from excitation lasers. This also affects the level of tissue penetration for 3D imaging, before photobleaching occurs. Two-photon microscopy addressed this issue by “splitting” a single photon into two photons with half the energy. This allowed comparable excitation while achieving significant reduction in photobleaching as well as enhanced resolution. This hypothesis was formulated by Maria Gopport- Mayer and was put to practical use by Winfried Denk in the 1990s by construction of the first two-photon excitation (2PE) laser confocal microscope.11
Though confocal and two-photon microscopy vastly improved the issues of resolution and photobleaching, they still proved inadequate for the purpose of single-molecule resolution and imaging. TIRF exploits the principle of reflection in such a way that the excitation beam is aimed at the critical angle that causes total internal reflection on the surface of the cover-glass. This assists in achieving enhanced resolution close to the surface of the cover-glass to a depth of 100 nm or less, thereby matching atomic scale resolution. The advantages of TIRF was successfully demonstrated with single ATP turn-over reaction in single myosin molecules in 1995.8
This post provides just a few glimpses into the vast repertoire of possibilities offered by fluorescence microscopy. To elaborate the variants of this rapidly growing field is beyond the scope of this article. But an important take-home point is the necessity of high-quality fluorophores suitable for both live and fixed specimen imaging. Biotium offers the widest range of reagents needed for all arenas of fluorescent imaging. Through cutting-edge research and development, Biotium is dedicated to developing high-performance tools for fluorescence-based biological research.

Gopal Ramakrishnan holds a PhD in Biochemistry from the All India Institute of Medical Sciences. Dr. Ramakrishnan received his Post-Doctoral training at the Cancer Institute, University of Mississippi Medical Centre where he studied cell signaling, metabolism, development and cancer biology. He currently serves as a Research Assistant Professor at the University of Illinois in Chicago.
