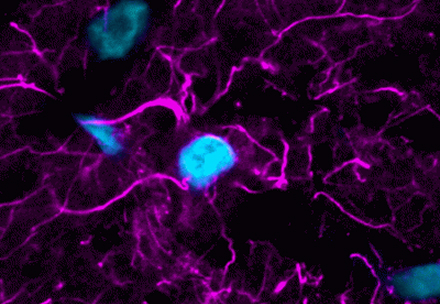
To learn more about western blotting, including the advantages of near-infrared fluorescence detection, see our webinar: Fundamentals of Western Immunoblotting: Chemiluminescence and NIR Multiplex Imaging (49:44).
Workflow overview:
- Optional: Perform total protein prestaining
- Perform SDS-PAGE and protein transfer (~2 hours) (optional stopping point)
- Optional: Confirm protein transfer
- Blocking (30 - 60 min.)
- Primary antibody incubation (2 hours or overnight)
- Washes (~30-60 min.)
- Secondary antibody incubation (not required for labeled primary antibody) (30 min. to 2 hours)
- Washes (~30-60 min.)
- Dry membrane (optional stopping point)
- Scan fluorescence
General considerations for fluorescent western detection:
- Multiplex fluorescence western detection requires an imaging system capable of detecting multiple fluorescent conjugates. For best results, use a gel imager or scanner specifically designed for imaging fluorescent blots.
- Far-red or near-infrared dyes are optimal for fluorescent western, because background is lower in these wavelengths. Visible fluorescent dyes can be used, but generally will have lower signal-to-noise ratio due to higher autofluorescence of proteins and blotting membranes in the visible spectrum.
- Optimal protein loading amount varies depending on detection method and target expression level, but ranges between 1-10 ug/lane for most applications.
- Blue tracking dyes in SDS-PAGE loading buffer can fluoresce in the far-red/near-infrared spectra; loading buffer with an orange tracking dye is recommended for fluorescent western detection.
- Either nitrocellulose or PVDF may be used for fluorescent western, but autofluorescence can vary widely among different sources of blotting membrane. In our experience, nitrocellulose and low fluorescence PVDF membranes show similar background fluorescence, but PVDF can give higher sensitivity, possibly due to higher protein binding.
- After protein transfer, dried blotting membranes can be stored at room temperature for months to years prior to detection.
- 9 cm2 petri dishes hold 5-10 mL and are convenient for washing and incubating mini-blots. Alternatively, commercially available black blotting boxes for fluorescent westerns come in a variety of sizes for blots or membrane strips.
- Before the development of chemiluminescence-based and fluorescence-based western detection, alkaline phosphatase substrates were commonly used for western detection. At that time, Tris-buffered saline (TBS) was the buffer of choice for western blots, because phosphate buffers could interfere with alkaline phosphatase signal development. In our experience, PBS and TBS can be used for routine fluorescent western detection with similar results. Some researchers prefer to use TBS for phosphoprotein detection out of concern that phosphate buffers may interfere with phospho-specific antibody binding.
- BSA, non-fat dry milk, and fish gelatin can be used for western blot blocking and antibody dilution buffers. These blocking agents are usually used at 1-5% in PBS (or TBS) + 0.1% Tween®20. Commercially available blocking buffers developed specifically for fluorescent western detection, such as our TrueBlack® WB Blocking Buffer, can give lower background than other blocking agents.
- It may be desirable to minimize the volume of antibody solutions used for blotting by using sealable bags or small containers. Enough solution should be used to freely move across the blot without trapping bubbles.
- For blocking and wash steps, don't skimp on volume. Use 5-10 mL buffer for a mini-blot. The blot should move freely in the buffer.
Procedure:
- Optional: To fluorescently label total protein in your sample for transfer confirmation and western normalization, use a total protein prestaining kit, such as our Mix-n-Stain™ Total Protein Prestain Kit, according the kit protocol.
- Perform SDS-PAGE and western transfer using standard protocols.
Note: After transfer, membranes can be rinsed in water, dried, and stored between sheets of filter paper at room temperature for months or longer.
- Optional: Confirm protein transfer by imaging total protein prestain (if used), or by staining the membrane with Ponceau S dye according to the supplier instructions.
Note: Ponceau S can be used for visual staining of cell lysate proteins at ~10 ug total protein per lane, but may not be sensitive enough to detect lower protein loading amounts. Our Mix-n-Stain™ Total Protein Prestain Kit can detect as little as 1 ng total protein per lane.
- If using PVDF membranes, re-wet the membrane in methanol, then rinse in water. For nitrocellulose membranes, proceed to step 5.
- Place blot in a clean dish containing blocking buffer of your choice. Use enough buffer to completely cover the blot and allow it to move freely in the dish.
- Block membrane for 30 min. to 1 hour at room temperature with gentle rocking.
- Dilute primary antibody to recommended concentration in fresh blocking buffer. Pour off the blocking buffer and add enough diluted antibody solution to allow the membrane to move freely with no stationary bubbles or dry spots.
- Incubate membrane with gentle rocking for 1-2 hours at room temperature or overnight at 4°C. If using fluorescently labeled primary antibodies, protect from light.
- Rinse membrane three times with PBS or TBS with 0.1% Tween®-20, then wash 5x for 5-10 min. each wash with rocking. Use a generous amount of wash buffer so blots move freely during washes.
If using fluorescently labeled primary antibodies, continue to step 11. If using labeled secondary antibody conjugates, continue to step 10.
- Dilute secondary antibody in fresh blocking buffer at the concentration recommended by the supplier for western blot (usually in the range of 50-100 ng/mL). Add to blot as in step 7. Incubate 30 min. to 2 hours with rocking.
Note: Some near-IR secondary antibody conjugates require additional detergent to be added to the buffer, check the supplier instructions for your antibody conjugate and blocking buffer for recommendations.
- Wash membrane as in step 9.
- Rinse blot once in buffer without detergent and dry before imaging using a compatible fluorescence imaging system.
Note: Dried blots can be stored between sheets of filter paper at room temperature in the dark and re-scanned after months or even years.
Note: Keep blots wet at all times and store in buffer if they are to be stripped and probed with additional antibodies.
 New Products
New Products Earth-Friendly Products
Earth-Friendly Products Biotium Choice Antibodies
Biotium Choice Antibodies Special Offers
Special Offers