 New Products
New Products Earth-Friendly Products
Earth-Friendly Products Biotium Choice Antibodies
Biotium Choice Antibodies Special Offers
Special Offers







Content #1
Content #1
Content #1
Working with RNA can be challenging and intimidating compared to working with DNA. While DNA is known to be quite stable and hardy (geneticists have been sequencing genomes that are thousands of years old), the chemical structure of RNA makes it prone to hydrolysis and, therefore, much less stable. And while nucleases can degrade both DNA and RNA, RNase enzymes are highly stable and readily refold after denaturation, making them difficult to inactivate permanently. Additionally, contamination is a very real concern when working with RNA. For this reason, it is crucial to use clean gloves and a clean lab coat, keep tubes and bottles closed, use a clean (RNase-free) work area, and ensure you use RNase-free supplies, solutions, and reagents. Finally, while there are many fluorescence-based reagents to quantify and visualize DNA, few are optimal for RNA due to low sensitivity or a lack of specificity in the presence of contaminating DNA.
In this Tech Tip, we will simplify and demystify RNA work, and recommend tools that will help you to confidently isolate and analyze your RNA samples with ease.
Good RNA hygiene starts with PPE (personal protective equipment). When working with RNA, it is important to always be aware of what you are touching with your gloves. Avoid touching your face or hair since that could allow transfer of RNase from your skin to your gloves. Change gloves if you have touched unclean equipment or surfaces. We particularly recommend always putting on fresh gloves before handling pure RNA samples.
Decontamination of surfaces and tools
Before beginning RNA work, it is a good idea to decontaminate your workspace, including the benchtop, your pipettes, and other equipment that you will be using. We recommend cleaning these surfaces by spraying them with RNase-X™ Decontamination Solution, wiping them down, then spraying them with either dH2O or 70% ethanol and wiping again. RNase-X™ is very efficient at removing RNases from surfaces (Fig. 1).
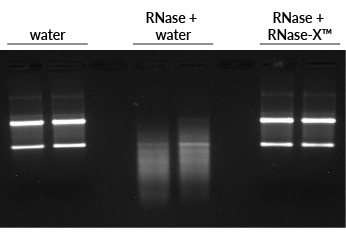
Figure 1. RNase-X™ eliminates RNase from surfaces. To simulate RNase-contaminated surfaces, the insides of microfuge tubes were coated with RNase A, then rinsed with either water or RNase-X™, followed by water washes. The final water sample was incubated with total human RNA, then run on an agarose gel with EMBER500™ RNA Prestain Loading Dye. First two lanes: RNA incubated with water alone shows bright ribosomal RNA bands. Middle two lanes: RNA incubated with the water from RNase-treated, water-rinsed tube is degraded. Last two lanes: RNA incubated with the water from RNase-treated, RNase-X™-rinsed tube is intact, like the control.
Tubes, tips, water, buffers, and reagents that will be used for RNA work must be RNase-free. Certified RNase-free products (such as Water, Ultrapure Molecular Biology Grade) can be purchased from many companies. Homemade solutions made with high-grade ingredients and ultrapure water are often RNase-free; this can be verified using RNase testing assays such as RNaseRevealTM Activity Assay Kit (see RNase testing section below). Another common method is to treat solutions with DEPC (diethyl pyrocarbonate) to inactivate RNases. However, DEPC treatment has several drawbacks: it can't be used with Tris-containing or other amino-containing solutions, it requires autoclaving, it may not fully inactivate RNases, it leaves the solution slightly acidic, and the DEPC by-products may inhibit downstream processes. So, we prefer to skip the DEPC and use solutions that have been verified as RNase-free.
Apart from enzymatic degradation caused by RNases, RNA is also susceptible to hydrolysis, which is an intramolecular reaction that causes breakage of the phosphodiester backbone. EDTA has a protective effect because divalent cations like Mg2+ increase hydrolysis. RNA hydrolysis is worse in alkaline solutions, so RNA should not be stored in a solution with pH higher than 7.5. We have seen good stability of RNA stored in either citrate buffer pH 6 or TE buffer pH 7.5 (Fig. 2). Temperature is the most important factor in RNA stability during storage: for best stability and for long-term storage, RNA should be kept at -70°C. For short-term storage, we have found RNA to be stable at 4°C or -20°C for at least 3 weeks (Fig. 2).
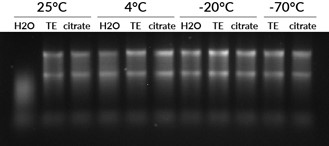
Figure 2. RNA stability depends on buffer and temperature. Total human RNA was suspended in either RNase-free water, TE (pH 7.5), or sodium citrate (pH 6). The samples were stored for 3 weeks at 25°C, 4°C, -20°C, or -70°C. The RNA stored at 25°C in water was degraded while the RNA stored at 25°C in TE or citrate buffer was still largely intact, showing the importance of storing RNA in buffer. However, RNA stored in water at 4°C or colder was still intact after 3 weeks, showing the importance of low temperature on RNA stability.
To keep RNA stabilized during handling or storage, products like RNAlaterTM can be added to tissues, cells, or lysates. Flash freezing or homogenization in TRIzol and freezing at -80°C are alternatives to products like RNAlaterTM. These stabilizing methods can offer flexibility to a researcher so that samples can be handled at room temperature, shipped, or treated to multiple freeze-thaw cycles without the RNA becoming degraded. However, any stabilization reagent added to a sample must be removed and the RNA purified before any downstream work is done.
RNase inhibitors, such as RiboGuardTM, may be helpful in some situations. These are proteins that inhibit specific types of RNases by binding to them in a 1:1 ratio. Their effectiveness depends on the amount of RNase contamination in a sample. Trace amounts of RNase may be effectively inhibited, but samples containing larger amounts of RNase (e.g., RNase-treated DNA samples) may not show full RNase inhibition. Some other caveats to using these RNase inhibitors are that they can be expensive, and their effects are non-covalent and reversible. Also, they contain glycerol and DTT, which may affect downstream applications. After attempting unsuccessfully to use RNase inhibitors to inactivate RNases in RNase-treated DNA, Biotium developed a proprietary method to permanently inactivate RNases in a solution, which we use to make RNase-Free Calf Thymus DNA.
When preparing solutions to use for RNA work (see Materials section above), it is useful to verify that the solution is free of RNase. With this in mind, Biotium developed the RNaseRevealTM Activity Assay Kit. This fluorescent assay was designed for the sensitive detection of RNase activity in liquid samples and is ideal for detecting RNase contamination in quality control workflows. The kit utilizes RNaseRevealTM Substrate, which is an RNA probe tagged with a green fluorophore and a quencher so that the intact probe is non-fluorescent. In the presence of RNase, the probe is cleaved, and the fluorophore is detached from the quencher, releasing a green fluorescent signal. Biotium also offers RNase A as a separate product that is recommended for use with this kit.
However, be aware that samples with extreme pH, high salt, or detergent can inhibit fluorescence-based RNase assays by directly affecting the fluorogenic RNase substrate or the RNase enzyme. This is easy to test for by adding RNase to your solution and ensuring that it registers fluorescence in the assay comparable to water plus RNase.
Isolation from fresh cells or tissues
For cultured cells and fresh tissues, total cellular RNA can be isolated by several different methods. The original method of extracting nucleic acids uses phenol and chloroform to separate proteins and lipids into an organic phase and nucleic acids into an aqueous phase. This process can be done under acidic conditions to selectively extract RNA from samples. A related method is to use TRI ReagentTM (also called TRIzolTM), which uses guanidinium salt in combination with phenol and chloroform to selectively isolate RNA.
These days, many researchers prefer to use column-based RNA isolation kits to avoid the use of phenol-based methods that involve hazardous chemicals and tricky techniques required to separate organic and aqueous phases. For those looking for a simpler purification solution, there are easy-to-use kits available such as the CELLDATA RNAstormTM Fresh Cell and Tissue RNA Isolation Kit. Many such kits use guanidinium salt and silica membrane columns to selectively isolate RNA, while others use magnetic beads.
Total RNA isolated from these methods consists of ~95% ribosomal RNA. For some applications, it may be desirable to purify mRNA from the total RNA. However, because mRNA makes up a small percentage of cellular RNA, it can be difficult to obtain high yields of mRNA, and to remove all of the ribosomal RNA. Methods involving beads coated with oligo dT that bind to the poly A tails of mRNA are most commonly used to enrich mRNA from a sample.
Isolation from FFPE tissue sections
With the advent of next-generation sequencing methods, researchers increasingly wish to isolate RNA from formalin-fixed paraffin-embedded (FFPE) tissues since patient samples are often preserved in this way. Purifying high quality RNA from FFPE tissues is more challenging than from fresh cells because formaldehyde chemically crosslinks the RNA to proteins and to itself. Some methods to reverse this cross-linking, such as high heat, can result in fragmented or poor-quality RNA.
The CELLDATA RNAstormTM 2.0 FFPE RNA Extraction Kit uses proprietary technology to chemically reverse formaldehyde cross-linking while avoiding the use of high temperatures and hazardous solvents. This leads to RNA with less fragmentation and greater amplifiability compared to other FFPE isolation kits, as measured by RIN score or by DV200 (Fig. 3). For high-throughput applications, we recommend the CELLDATA RNAstormTM 2.0 MagBead FFPE RNA Extraction Kit. This kit uses magnetic beads for RNA isolation and may be adapted for a multiwell plate format.
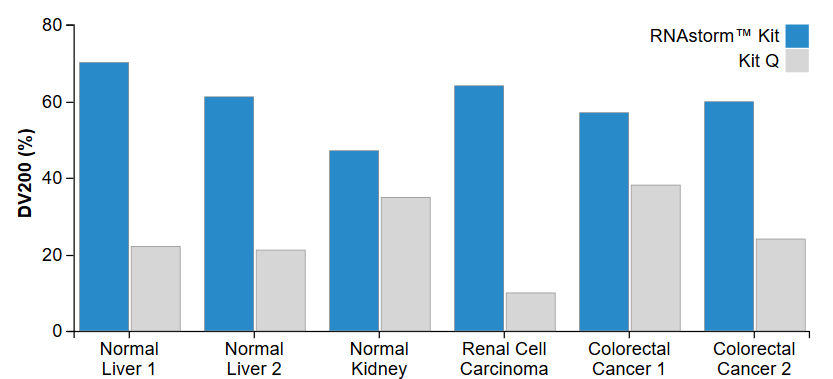
Figure 3. Increased DV200 values are observed for RNA extracted using the RNAstorm™ kit relative to a popular commercial kit. The DV200 represents percentage of RNA with length greater than 200 nucleotides, as measured using an Agilent Bioanalyzer RNA 6000 Nano Kit.
Isolation from extracellular vesicles
The contents of exosomes and other extracellular vesicles (EVs) have garnered increasing attention over recent years for their potential applications in both diagnostics and therapeutics. This includes RNA and its potential role as a key biomarker for cancer and other diseases. However, obtaining high yields of quality RNA from EVs is a challenge due to the dilute nature of EV samples and low RNA content within EVs. Additionally, short non-coding RNAs within EVs, such as miRNAs, often require specialized methods for efficient extraction.
The scientists at Biotium developed the ExoBriteTM EV Total RNA Isolation Kit with these challenges in mind, creating an easy-to-use kit for total RNA isolation (including mRNA and miRNA) from purified EVs. The RNA isolation procedure is a simple column purification method that takes as little as 20 minutes and requires no phenol/chloroform or ethanol precipitation steps. The recovery is estimated to be ~10-20 ng of RNA from 1×1010 SEC-enriched EVs. Please note that this amount is primarily dependent on the number and quality of EVs, which may be affected by purification methods, storage conditions, and the number of freeze-thaws. The isolated EV RNA can then be used for downstream analysis such as qPCR or RNAseq.
The classic method of quantifying nucleic acids is by measuring UV absorbance at 260 nm (A260). Although A260 nm will not distinguish between RNA and DNA, it can be a useful way to quantify your RNA if you are certain that it is very pure (i.e., that there are no free nucleotides or DNA in your sample). For RNA, multiplying the A260 nm by 40 gives the concentration in ng/uL. It is typical to also take the absorbance at 280 nm to look for contaminants like protein that absorb strongly at 280 nm; a 260/280 ratio of 2.0 or higher is generally considered to be pure RNA. However, be aware that the 260/280 ratio can vary depending on pH. We recommend always using the same solvent, if possible, to make your absorbance readings consistent between experiments (we always use TE pH 7.5 to control pH for consistency).
Fluorescent dye-based RNA quantitation assays are the most accurate way to quantify RNA because they have much greater sensitivity and linearity than A260 measurement. In addition, some (but not all) assays also have the advantage of being selective for ssRNA over dsDNA so that you can achieve an accurate RNA concentration even in the presence of contaminating DNA. The AccuBlue® Broad Range RNA Quantitation Kit is highly selective for RNA over dsDNA (Fig. 4) and is accurate over a wide range of RNA concentrations. Be aware that some other companies fluorescent RNA detection dyes, such as RiboGreen®, while sensitive, are not RNA-specific and will also detect dsDNA present in the sample.
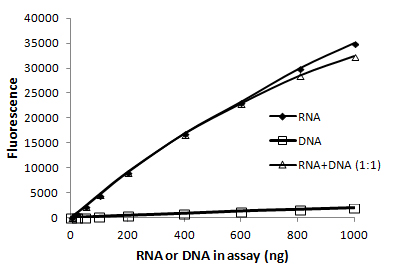
Figure 4. The AccuBlue® Broad Range RNA quantitation assay is highly selective for RNA over dsDNA. The presence of an equal amount of dsDNA in the sample has a negligible effect.
You may want to perform gel electrophoresis to determine the integrity or size of your RNA. Researchers often use the TapeStation system, Bioanalyzer RNA Analysis, or Fragment Analyzer systems to look at integrity and get a size profile instead of gels, especially for high-throughput experiments. It is often assumed that RNA electrophoresis always requires toxic, complicated formaldehyde or glyoxal gels to denature the RNA so that secondary structure doesnt affect the migration through the gel. However, we find that adding denaturants to the agarose gel is not necessary for evaluating RNA integrity. If your aim is just to visually evaluate your RNA, a regular agarose gel can be used as long as a denaturation agent is included in the loading buffer.
For increased sensitivity, convenience, and ease, Biotium created the EMBERTM Ultra RNA Gel Kit, a highly sensitive RNA gel stain available in a convenient pre-coated agarose format that includes formamide-containing RNA loading dye (Fig. 5). In addition to being far more sensitive than other fluorescent RNA gel stains currently available, EMBERTM Ultra does not require a post-electrophoresis staining step. This allows for a streamlined process of simply casting the agarose gel using the EMBERTM Ultra Precoated Agarose and then preparing the samples in the denaturing EMBERTM Ultra RNA Loading Dye. Bands can be imaged immediately after electrophoresis. RNA gel electrophoresis can also be done using polyacrylamide (PAGE) gels if you are looking at miRNA or other RNA up to 200 nucleotides long. Please note that EMBERTM Ultra is not compatible with PAGE gels. These gels require the use of a denaturing loading buffer that does not contain fluorescent dye and post-staining with a sensitive nucleic acid dye like Oxazole Gold.
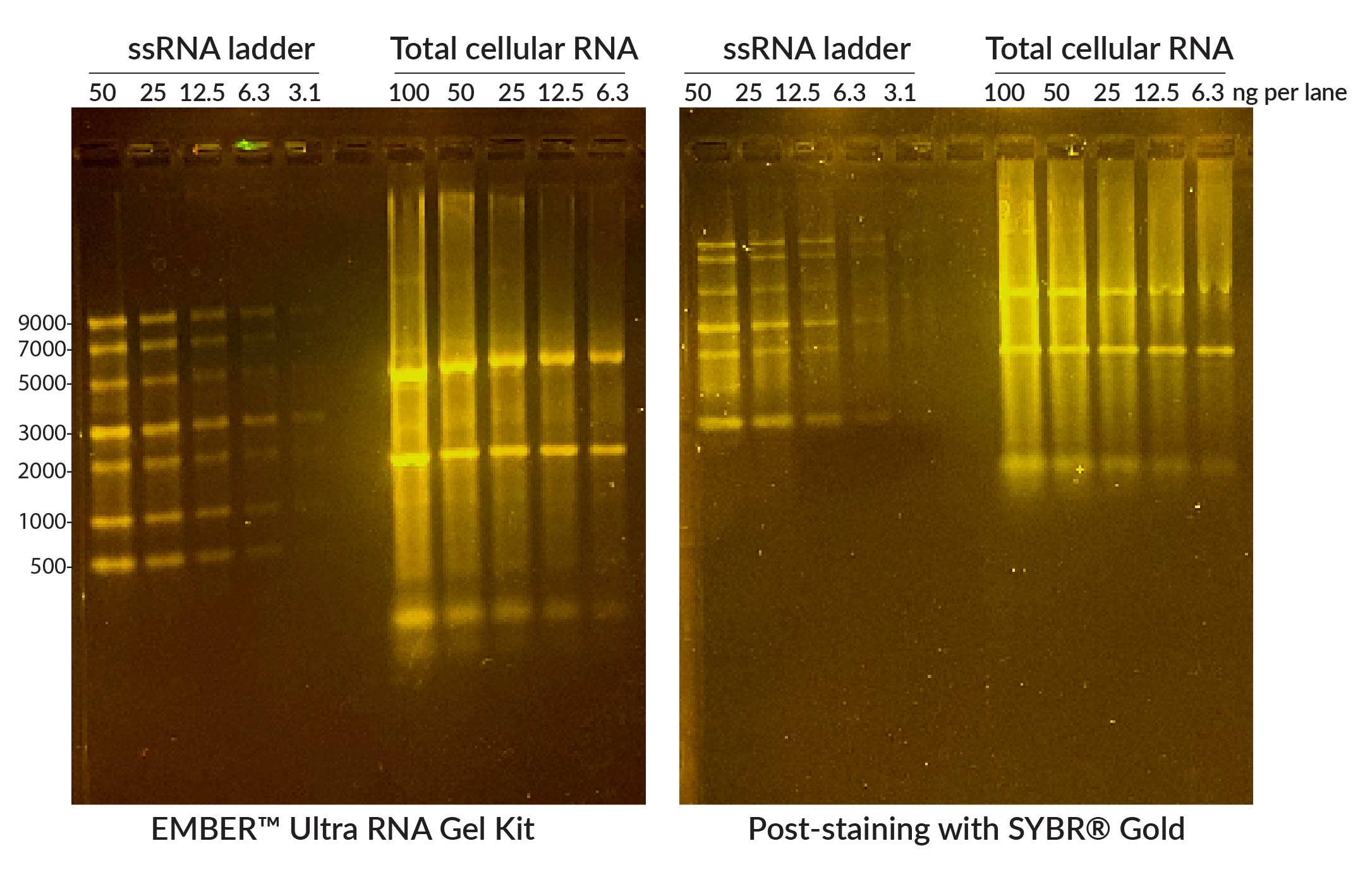
Figure 5. Comparison of EMBERTM Ultra RNA Gel (left) with 1% agarose gel post-stained with SYBR® Gold. NEB ssRNA Ladder or total Jurkat cell RNA were denatured at 70°C for 10 minutes in EMBERTM Ultra RNA Loading Dye (for EMBERTM gel) or NEB RNA Loading Dye (for SYBR® Gel) and run on 1% agarose/1X TBE gels. EMBER® Ultra Gel was imaged immediately after electrophoresis, while the other gel was post-stained in 1X SYBR® Gold/1X TBE for 30 minutes. Gels were imaged on Biotium's Gel-BrightTM Laser Diode Gel Illuminator. The EMBER Ultra® RNA Gel Kit offers comparable or superior sensitivity and resolution compared to SYBR® Gold, without a time-consuming post-staining step.

Content #1
Content #1
Content #1
Content #2
Content #3