New Products
New Products Earth-Friendly Products
Earth-Friendly Products Biotium Choice Antibodies
Biotium Choice Antibodies Special Offers
Special Offers
CF® Dye Antibodies and Other Conjugates
Primary Antibodies
- Growing collection of more than 2000 monoclonal antibodies
- Validated in IHC and other applications
- Choose from 6 bright and photostable CF® Dyes or biotin
- Available BSA-free, ready to use for Mix-n-Stain™ labeling
- Affordable 100 uL sizes available
Secondary & Anti-Tag Antibodies
- Secondary and anti-tag antibodies
- >20 CF® Dye colors, R-PE, APC, HRP, AP or biotin
- Highly cross-adsorbed F(ab’)2 fragments and isotype-specific options
- Single label CF® Dye conjugates for super-resolution STORM
- Affordable 50 uL sizes available

Primary Antibodies – sizes and formats:
| Format | Concentration | Size |
|---|---|---|
| CF® Dye conjugates (6 colors) | 0.1 mg/mL | 100 uL or 500 uL |
| Biotin conjugates | 0.1 mg/mL | 100 uL or 500 uL |
| Purified, with BSA | 0.2 mg/mL | 100 uL or 500 uL |
| Purified, BSA-free (Mix-n-Stain™ Ready) | 1 mg/mL | 50 uL |
Secondary Antibodies – sizes and formats:
| Format | Concentration | Size |
|---|---|---|
| CF® Dye and biotin conjugates | 2 mg/mL | 50 uL, 500 uL, or 1 mg |
| HRP conjugates | 1 mg/mL | 100 uL, 1 mL, or 1 mg |
| AP conjugates | 1 mg/mL | 100 uL or 1 mg |
| R-PE conjugates | 0.5 mg/mL | 200 uL or 1 mL |
| APC conjugates | 0.5 mg/mL | 100 uL or 500 uL |
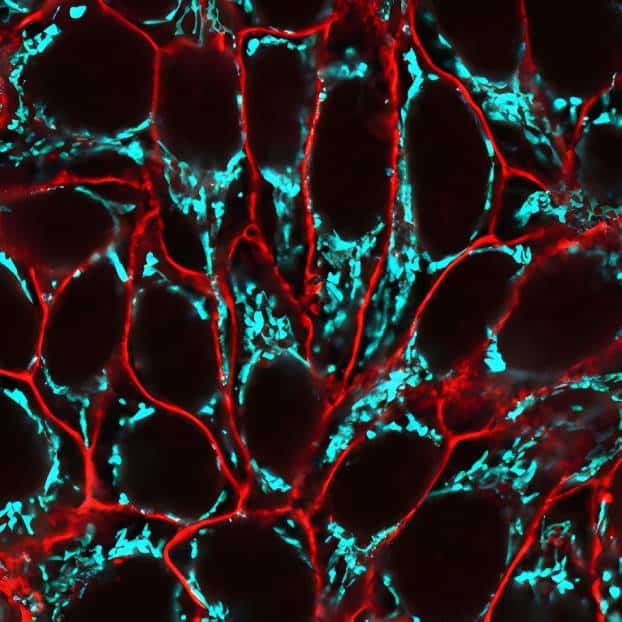
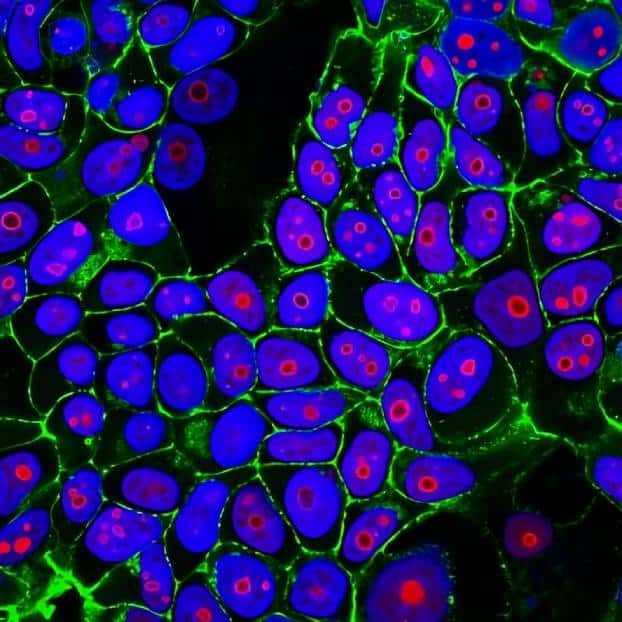
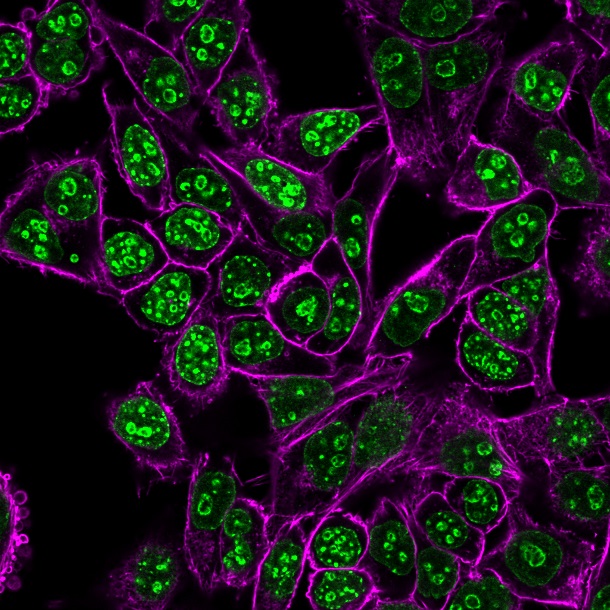
CF® Dye Bioconjugates
We offer several CF® Dye bioconjugates suitable for immunofluorescence workflows.
- Annexin V, nucleotide conjugates, and TUNEL assays for studying apoptosis
- Glycoprotein probes and cell surface stains: ConA, WGA, and PNA lectins
- Phalloidin conjugates for microtubule staining
- Streptavidin and biotin conjugates, and more
Antibody Labeling Kits
- Label in only 15 minutes
- Labeling Kits for 32 CF® Dyes, FITC, Biotin, R-PE, APC, and more
- Stable covalent labeling, no purification required
- Compatible with common antibody stabilizers
- Kits designed for Nanobodies® (ie. camelid single variable or VHH domains) available
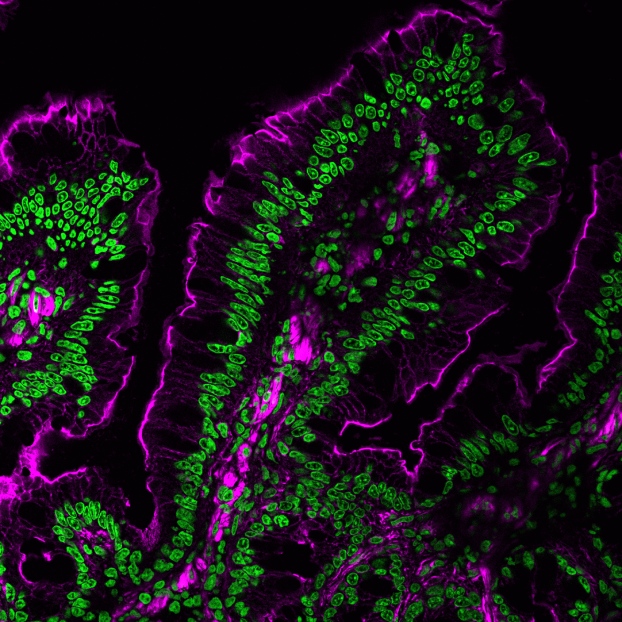
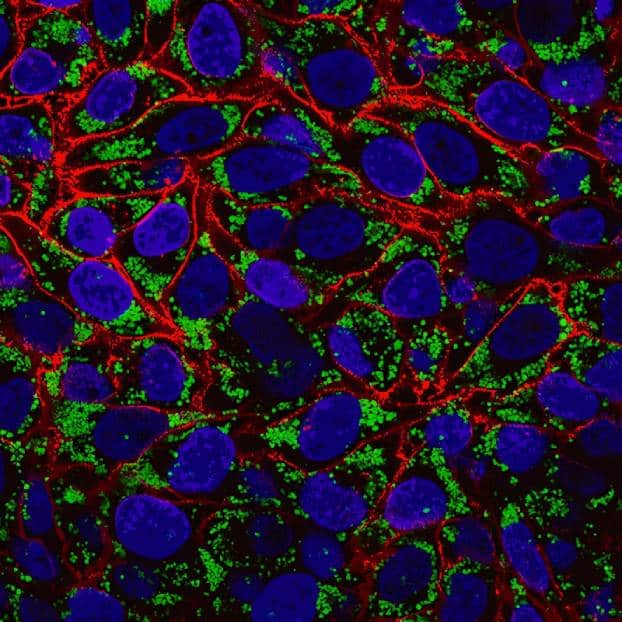
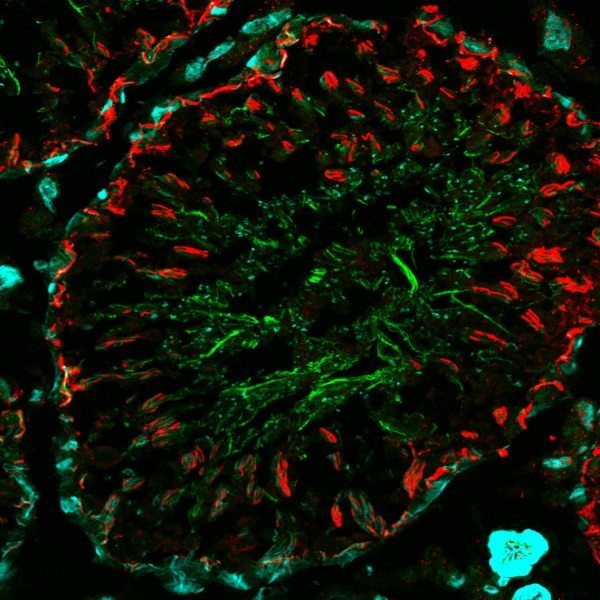
CF® Dyes for Super-Resolution Applications

Looking for organelle specific stains? View our Cellular Stains page for a comprehensive list of products specifically designed for staining nucleus, cytoplasm, mitochondria, membranes, lysosomes, and other cellular components.
Also, view our Cellular Stains Comparison Guide for a quick reference on targets, applications, fixability, staining in different organisms, and more information on our cell stains. This guide will help you ensure that you choose the right dye for your specific application.
Tyramide Signal Amplification
Tyramide signal amplification (TSA) is a highly sensitive method enabling the detection of low-abundance targets in fluorescent immunocytochemistry (ICC), immunohistochemistry (IHC), and in situ hybridization (FISH) applications. TSA involves horseradish peroxidase (HRP)-catalyzed deposition of tyramide on and near a target protein or nucleic acid sequence in situ.
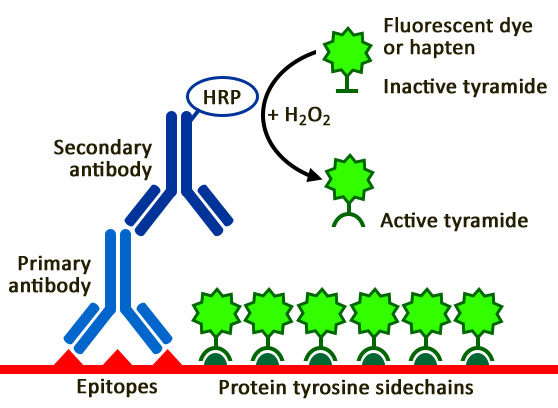
Tyramide Signal Amplification Kits
- Our kits provide all critical reagents for tyramide labeling
- Choose your tyramide: biotin tyramide or one of 6 CF® Dye tyramides
- Choose your HRP conjugate: goat anti-mouse, goat anti-rabbit, or streptavidin
- Kits also include Amplification Buffer and BSA for blocking
Tyramide Amplification Buffers
- Tyramide Amplification Buffer Plus has enhanced sensitivity for TSA, with improved brightness, specificity, and sensitivity over our original buffer below.
- Ready-to-Use Tyramide Amplification Buffer was our original buffer, and has been replaced with the above buffer. The advantage of this buffer is that there is no need to add hydrogen peroxide.
Tyramide Signal Amplification Kits
| Tyramide Label | Ex/Em | Secondary conjugate | Catalog no. |
|---|---|---|---|
| CF®488A | 490/515 nm | Goat anti-mouse HRP | 33000 |
| Goat anti-rabbit HRP | 33001 | ||
| Streptavidin HRP | 33002 | ||
| CF®543 | 541/560 nm | Goat anti-mouse HRP | 33003 |
| Goat anti-rabbit HRP | 33004 | ||
| Streptavidin HRP | 33005 | ||
| CF®568 | 562/583 nm | Goat anti-mouse HRP | 33006 |
| Goat anti-rabbit HRP | 33007 | ||
| Streptavidin HRP | 33008 | ||
| CF®594 | 593/614 nm | Goat anti-mouse HRP | 33009 |
| Goat anti-rabbit HRP | 33010 | ||
| Streptavidin HRP | 33011 | ||
| CF®640R | 642/662 nm | Goat anti-mouse HRP | 33012 |
| Goat anti-rabbit HRP | 33013 | ||
| Streptavidin HRP | 33014 | ||
| CF®680R | 680/701 nm | Goat anti-mouse HRP | 33015 |
| Goat anti-rabbit HRP | 33016 | ||
| Streptavidin HRP | 33017 | ||
| Biotin-XX | N/A | Goat anti-mouse HRP | 33018 |
| Goat anti-rabbit HRP | 33019 | ||
| Streptavidin HRP | 33020 |
Standalone Dye and Hapten Labeled Tyramides
| Tyramide label | Ex/Em | Size | Catalog no. |
|---|---|---|---|
| CF®350 | 347/448 nm | 0.5 mg | 92170 |
| CF®405L | 395/545 nm | 0.5 mg | 92198 |
| CF®405S | 404/431 nm | 0.5 mg | 92197 |
| CF®405M | 408/452 nm | 0.5 mg | 96057 |
| CF®430 | 426/498 nm | 0.5 mg | 96053 |
| CF®488A | 490/515 nm | 0.5 mg | 92171 |
| FITC | 492/514 nm | 0.5 mg | 96018 |
| CF®514 | 516/548 nm | 0.5 mg | 92199 |
| CF®532 | 527/558 nm | 0.5 mg | 96066 |
| CF®543 | 541/560 nm | 0.5 mg | 92172 |
| CF®550R | 551/577 nm | 0.5 mg | 96077 |
| CF®555 | 555/565 nm | 0.5 mg | 96021 |
| Cyanine 555 | 555/565 nm | 0.5 mg | 96020 |
| CF®568 | 562/583 nm | 0.5 mg | 92173 |
| CF®583R | 586/609 nm | 0.5 mg | 96085 |
| CF®594 | 593/614 nm | 0.5 mg | 92174 |
| CF®620R | 617/639 nm | 0.5 mg | 92194 |
| CF®640R | 642/662 nm | 0.5 mg | 92175 |
| CF®647 | 650/665 nm | 0.5 mg | 96022 |
| CF®660R | 663/682 nm | 0.5 mg | 92195 |
| CF®680R | 680/701 nm | 0.5 mg | 92196 |
| CF®710 | 712/736 nm | 0.5 mg | 96127 |
| CF®725 | 729/755 nm | 0.5 mg | 96128 |
| CF®740 | 742/767 nm | 0.5 mg | 96124 |
| CF®750 | 755/779 nm | 0.5 mg | 96052 |
| CF®754 | 748/793 nm | 0.5 mg | 96090 |
| Biotin-XX | N/A | 0.5 mg | 92176 |
| DNP | N/A | 0.5 mg | 96019 |
AntiFix™ Antigen Retrieval Buffer
Features
- Excellent antigen retrieval for a wide variety of targets at neutral pH
- Simplifies HIER optimization
- Validated with fluorescence staining using tyramide amplification
- Non-toxic and non-flammable
View Product Page

TrueBlack® Background Reducers
TrueBlack® Lipofuscin Autofluorescence Quenchers
Lipofuscin autofluorescence in certain human or aged animal tissues fluoresce brightly in all channels, making immunofluorescence virtually impossible unless lipofuscin fluorescence is masked. Traditionally, Sudan Black B has been used to quench lipofuscin autofluorescence but also introduces non-specific red and far-red fluorescence, limiting the use of fluorescent dyes in those wavelengths. Biotium’s line of TrueBlack® lipofuscin autofluorescence quenchers are a superior alternative to Sudan Black B to quench autofluorescence with much lower background. Learn more about our TrueBlack® Background Reducers.
TrueBlack® Plus: A Unique Lipofuscin Quencher
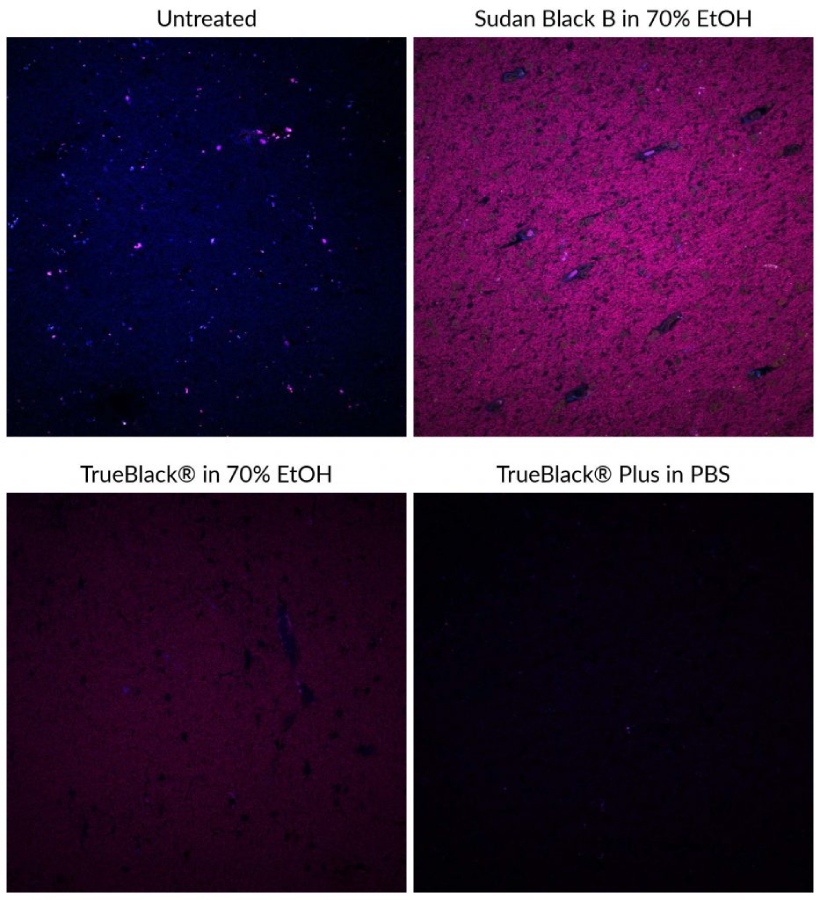
View Product Page
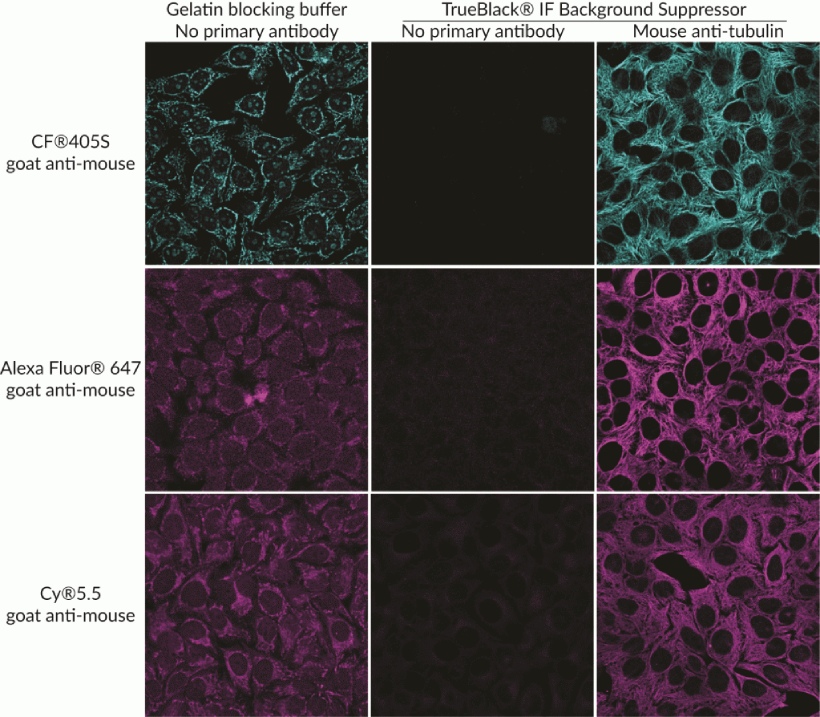
TrueBlack® IF Background Suppressor
The TrueBlack® Background Suppressor System is a buffer system designed for optimal blocking of non-specific staining for immunofluorescence (IF). The buffers are designed to block background from both non-specific antibody binding as well as direct interaction of fluorescent dyes on antibodies with cells or tissue sections.
Features
- Suppresses background from non-specific antibody binding and charged dyes
- More efficient than Image-iT® FX, block and permeabilize in just 10 minutes
- Complete system for blocking, permeabilizing, and antibody dilution
- Non-mammalian blocking agents, for broad secondary antibody compatibility
- For cells or tissue sections
View Product Page
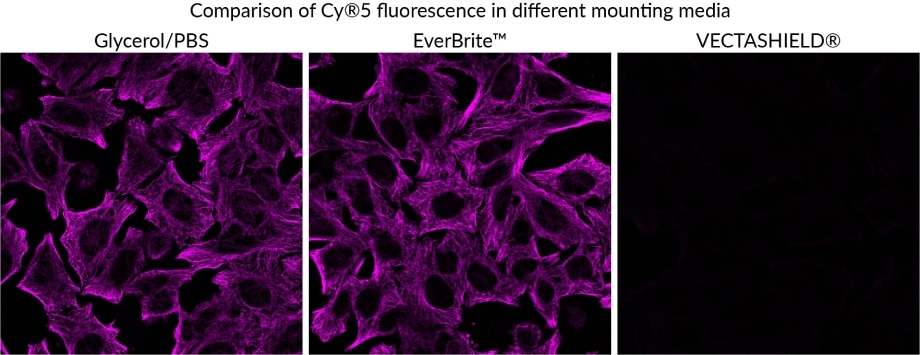
EverBrite™ Mounting Medium
Features
- Antifade mounting medium to prevent photobleaching of fluorophores
- Refractive index is well-matched to that of coverslip glass and immersion oil
- Available in wet-set or hardset formulations
- Available with DAPI, far-red NucSpot® 640, or without counterstain
- EverBrite TrueBlack® Hardset is the only mounting medium that quenches lipofuscin background

| Product | Nuclear Counterstain | Cat. No. | Features |
|---|---|---|---|
| EverBrite™ Mounting Medium | None | 23001 | • Wet-set mounting medium • Requires coverslip sealing • Refractive index 1.46 |
| EverBrite™ with DAPI | DAPI | 23002 | |
| Drop-n-Stain EverBrite™ Mounting Medium | None | 23008 | • Wet-set mounting medium • Convenient dropper bottle • Ideal for wells & chambers • Refractive index 1.42 |
| Drop-n-Stain EverBrite™ with DAPI | DAPI | 23009 | |
| EverBrite™ Hardset Mounting Medium | None | 23003 | • Hard-set mounting medium • Forms hard seal after 24 h • No coverslip sealing needed • Refractive index 1.42 after 24 h of curing, and 1.46 four days after curing |
| EverBrite™ Hardset with DAPI | DAPI | 23004 | |
| EverBrite™ Hardset with NucSpot® 640 | NucSpot® 640 | 23016 | |
| EverBrite TrueBlack® Hardset Mounting Medium | None | 23017 | • Unique antifade with lipofuscin quenching • Quenches as it hardens, with low background • Refractive index 1.42 after 24 h of curing, and 1.46 four days after curing |
| EverBrite TrueBlack® Hardset with DAPI | DAPI | 23018 | |
| EverBrite TrueBlack® Hardset with NucSpot® 640 | NucSpot® 640 | 23019 | |
| CoverGrip™ Coverslip Sealant | N/A | 23005 | • For sealing edges of wet-set coverslips |
Buffers and Other Accessories
CoverGrip™ Coverslip Sealant
CoverGrip™ Coverslip Sealant is the first product designed for sealing edges of wet-mounted coverslips. Unlike nail polish, CoverGrip™ contains no ingredients that can leach into aqueous mounting medium.
Features
- Seals edges of wet-mounted coverslips
- Dries hard and clear
- Less damaging to samples than nail polish
- Won’t leach into aqueous mounting medium and affect fluorescence
- Contains natural and environmentally friendly d-limonene

View Product Page
SuperHT Pap Pen
The SuperHT PAP Pen 2.0 draws a water repellent circle around slide mounted tissue and prevents the waste of valuable reagents by keeping the liquid contained in a single droplet. Our SuperHT PAP Pens are available in regular size (~800 applications), or mini size (~400 applications).
Features
- Draw water repellent barriers around tissue samples on slides
- Conserve precious antibodies and reagents
- Barriers are insoluble to aqueous buffer, detergents, alcohols, and acetone
- Barriers are stable up to 120°C

View Product Page
Other Products Useful for Immunofluorescence Microscopy
| Product | Catalog No. | Size | Features |
|---|---|---|---|
| Fixation Buffer | 22015 | 100 mL | • Ready-to-use formaldehyde-based fixation buffer |
| Permeabilization Buffer | 22016 | 100 mL | • Ready-to-use permeabilization buffer for intracellular staining |
| Permeabilization and Blocking Buffer (5X) | 22017 | 100 mL | • Concentrated buffer for one step permabilization and blocking for intracellular immunofluorescence • Goat serum-based blocking buffer |
| Flow Cytometry Fixation/Permeabilization Kit | 23006 | 50 tests | • Optimally formulated buffers for fixation/permabilization for intracellular staining for flow |
| 10X Fish Gelatin Blocking Agent | 22010 | 100 mL | • Provides excellent blocking for immunofluorescence or western • Add to buffer of your choice (PBS or TBS) • Compatible with goat and sheep primaries, unlike BSA, milk, or goat serum |
| Fish Gelatin Powder | 22011 | 2 x 50 g | • Gelatin from cold water fish skin for blocking for immunofluorescence or western • Compatible with goat and sheep primaries, unlike BSA, milk, or goat serum |
| Bovine Serum Albumin, 30% Solution | 22014 | 100 mL | • Commonly used blocking agent and antibody or protein stabilizer • 30% solution in water • Made from IgG-free, protease-free Fraction V BSA |
| Bovine Serum Albumin Fraction V | 22013 | 50 g | • Commonly used blocking agent and antibody or protein stabilizer • IgG-free, protease-free Fraction V BSA |
| Tween®-20 | 22002 | 50 mL | • Detergent commonly used for western blocking and washing |
| Mini Cell Scrapers | 22003 | Pack of 200 | • For harvesting cells or cell lysates from 96-, 48- and 24-well plates • 0.5 cm (3/16") wide and 6 cm (2 3/8") long • 20 packs of 10 scrapers per pack • Polyethylene, disposable & sterile |